1. Current Research and Principal Research
Interests
Research
Interests
My principal research interests
center on studies of genuinely organic molecule-based magnetism, and I
strive to develop novel magnetic materials such as single-component
organic "ferrimagnets" by quantum-chemistry-based crystal engineering
and to unravel the underlying mechanism for inter- and intramolecular
magnetic interactions in crystalline solid states on a quantum
mechanics basis.
The last decades have witnessed a
rapid development of molecule-based magnetism. In inorganic
transition metal-based magnets, materials with fascinating electrical
and optical properties have been developed. On the other hand, a
purely organic molecule-based ferromagnet with a three-dimensional
long-range ordering of unpaired electron spins has been a long-standing
issue in terms of materials challenge in molecular science and
chemistry. The first example of organic ferromagnet was
discovered in 1991 (M.
Tamura et al., Chem. Phys. Lett., 186,
401(1991)); I was
engaged in this discovery in my graduate school days. Since that
discovery, more than 30 ferromagnets have been well-documented in
genuinely organic molecule-based materials. Unpaired electrons
occupy molecular orbitals made up of s or p atomic orbitals in these
materials. This is in a remarkable contrast to traditional,
naturally found magnetic materials, which are atom-based, having d- or
f-orbital spin sites. My research interests lie in such
molecule-based magnets. The research is oriented towards basic
molecular sciences based on quantum chemistry, which include syntheses
of novel open-shell molecules, characterization of the molecules with
magnetic resonance spectroscopy (ESR) and solid-state magnetic
properties measurements, and quantum statistical or quantum chemical
calculations. These studies elucidate the mechanism of
intermolecular magnetic interactions and the relation between chemical
structures and magnetic functionalities of molecules or molecular
assemblages in solid states.
In organic magnets including
the above-mentioned ferromagnet, electron spins are spread over many
atomic sites in constituent open-shell molecules. Thus,
intermolecular magnetic interactions for organic magnets inherently
have a multicentered nature. This multicentered nature can be
negligible in traditional atom-based magnets and thus has been
overlooked historically so far. In 1997, I pointed out in quantum
terms, for the first time, that in genuinely organic molecule-based
magnetics an essential feature is multicentered magnetic interaction
within and between the molecules with two or more unpaired electrons,
i.e., S > 1/2. An
antiparallel coupling of different spin
quantum numbers, e.g., S = 1 and S = 1/2 has been
believed, without any
rationale, to give a ferrimagnetic spin alignment as schematically
shown in Fig.1a. A purely organic molecule-based ferrimagnet,
however, has not been discovered yet, which I have attributed to its
multicentered nature from theoretical calculations based on the spin
Hamiltonian as schematically shown in Fig.1b. I have been
examining the ferrimagnetic spin alignment in organic heteromolecular
systems from both theoretical and experimental sides. I strive
towards the development of new organic magnets while at the same time
unraveling the underlying mechanism for magnetic interactions on the
basis of quantum mechanics.
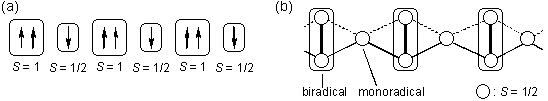
Fig. 1.
Schematic drawings of the heteromolecular assemblage as a model for
organic molecule-based ferrimagnetics (a) and the Heisenberg spin
Hamiltonian (b).
Current
Research
My current research interests fall into the following four issues.
(a)Theoretical Elucidation of
the Nature of Molecule-Based Ferrimagnetism. Preceding theoretical and
experimental studies on ferrimagnets are based on the simple picture in
Fig.1a. I have drawn attention to a magnetic degree of freedom
remaining in S > 1/2 molecules; this degree of freedom has
been
overlooked in the history of molecule-based magnetism. From
numerical calculations of expectation values for the biradical spin
<Sb2> with the model spin Hamiltonian of biradical-monoradical
alternating chain (Fig.1b), a spin contraction is found to occur at the
biradical: The <Sb2> value deviates from the value of Sb =
1 as expected for isolated molecules. An S = 1 is not a
good
quantum number for describing the biradical embedded in the molecular
assemblages. The contraction is the consequence of the internal
magnetic degree of freedom in the S > 1/2 molecule with
multicentered intermolecular interactions. Experimental evidence
for the internal magnetic degree of freedom has been found from ESR
spectra of an organic biradical embedded in a hetero-spin molecular
assemblage.
I have shown generation of an effective S = 1/2 spin in
the
alternating chain of biradicals and monoradicals with a Heitler-London
formulation. Molecular dimer of a biradical and a monoradical, or
the unit cell of the alternating chain, behaves as a magnetic
supramolecule with S = 1/2. Spin polarization of the
magnetic
supramolecules has been shown to bring about effectively ferromagnetic
interactions between the supramolecules as schematically shown in
Fig.2. Thus, the ferrimagnetic spin alignment in the
heteromolecular chains is shown to be equivalent to the ferromagnetic
alignment of the effective S = 1/2 spins. This
equivalence has
been explicitly derived for the first time in quantum terms. Very
recently, the equivalence was examined in a real open-shell molecular
system of an organic triradical in terms of thermodynamic properties
reflecting the magnetic degree of freedom.
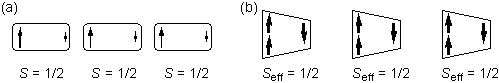
Fig.2.
Schematic diagrams for the ferromagnetic chain of spin polarized S
= 1/2 radicals (a) and the ferrimagnetic chain of spin-polarized
supramolecules with Seff = 1/2 composed of a
biradical and a monoradical (b). The arrows indicate the positive or
negative spin polarization.
(b)Single-Component Organic
Molecule-Based Ferrimagnetics. A practical difficulty in
constructing organic molecule-based ferrimagnetics is
co-crystallization of two kinds of molecules with different spin
quantum numbers S’s, e.g., S
= 1 and S = 1/2. As a purposive
molecular design for co-crystallizing distinct open-shell entities, I
have proposed a strategy of "single-component ferrimagnetics". An
organic triradical 1 (Fig.3a) has been designed and
synthesized, which
is composed of a π-biradical with a triplet (S = 1) ground state
and a π-radical with S = 1/2. In the triradical, the
π-conjugation between the S = 1 and the S=1/2 moieties
is
substantially truncated, giving a weakly coupled composite
π-system with two kinds of magnetic degree of freedom for S =
1 and S = 1/2 spins. An alternating chain of the S
= 1 and the S =
1/2 moieties has been found in 1 from an X-ray crystal
structure
analysis (Fig.3b and Fig.3c). Magnetic susceptibility of 1
has
indicated the occurrence of ferrimagnetic spin alignment in the
alternating chain. Thus, the triradical 1 is the first
example of
an organic molecule with the σ-bonded composite
π-system exhibiting a ferrimagnetic behavior. Syntheses
and magnetic characterization for triradicals 2, 3, and
4 (Scheme 1)
are in progress
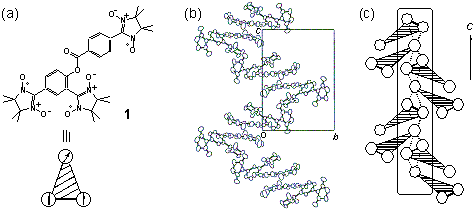
Fig.3. (a)
Triradical 1 with a schematic drawing of the three-spin system.
(b) Molecular packing of 1. (c) Schematic diagram of the chain.
The dashed lines represent the intermolecular interactions between the
biradical and the monoradical moieties along the chain.
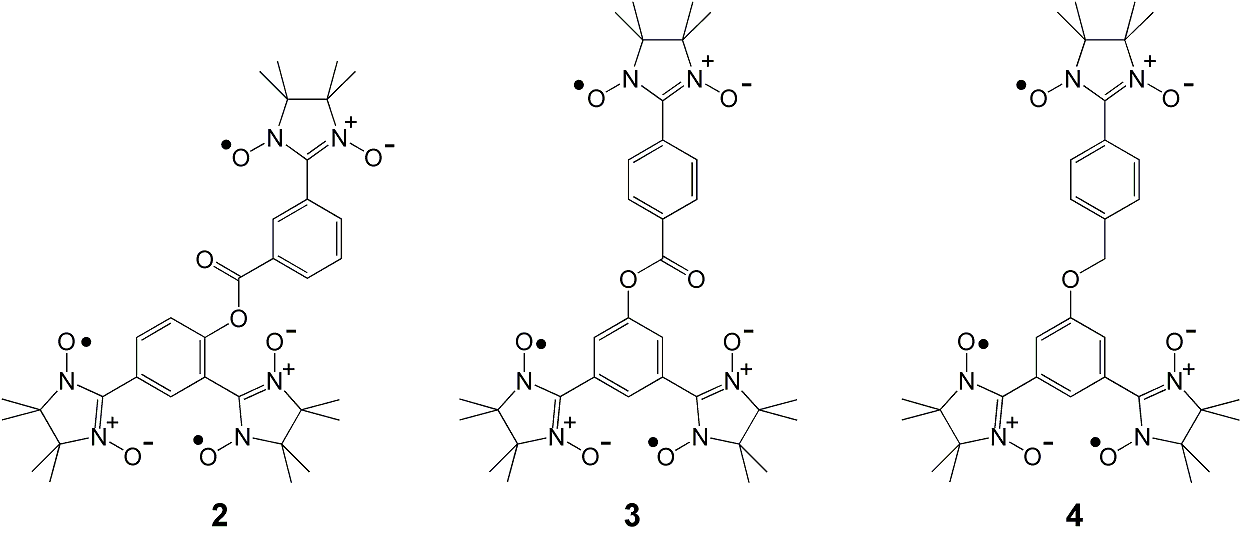
Scheme 1.
Triradicals as building blocks of
single-component ferrimagnetics.
(c) Supramolecular
Ferrimagnetics and Bio-Inspired Magnetics. Noncovalent bonding such
as hydrogen bonding and coulombic interaction between ionic charges can
be a promising driving force for crystallization of open-shell
molecules with differing S's, as shown in Scheme 2(a,b). We
have
designed and synthesized nitronyl nitroxide biradical with a pyridine
substituent 5 as an S = 1 component for the
supramolecular
ferrimagnets. The molecular ground states of the neutral biradical 5
and the cationic biradical 6+ were found to be
triplet (S
= 1)
from
magnetic susceptibility measurements, indicating that both 5
and 6+
serve as building blocks for organic supramolecular ferrimagnets. The
neutral biradical 5 was found to co-crystallize with p-benzoic
acid
substituted monoradical 7. The hydrogen-bonded complex 5-7
undergoes an
antiferromagnetic phase transition at 5 K.
Another type of hydrogen bonding-based molecular complexation is
pairing of nucleobases such as cytosine and guanine as found in DNA.
Nitronyl nitroxide radicals substituted with cytosine and guanine
bases, as shown in Scheme 2c, have been synthesized. The
hydrogen-bonded assemblage of the nucleobase-substituted radicals
uncovers a new category of bio-inspired molecule-based magnets and
bionano-architecture. This is termed “bio-inspired magnetics”. Studies
on open-shell molecular assembly which is templated by single stranded
oligonucleotides are in progress. The DNA-templated architecture is a
prototype of spin-mediated molecular/quantum information processing
devices.
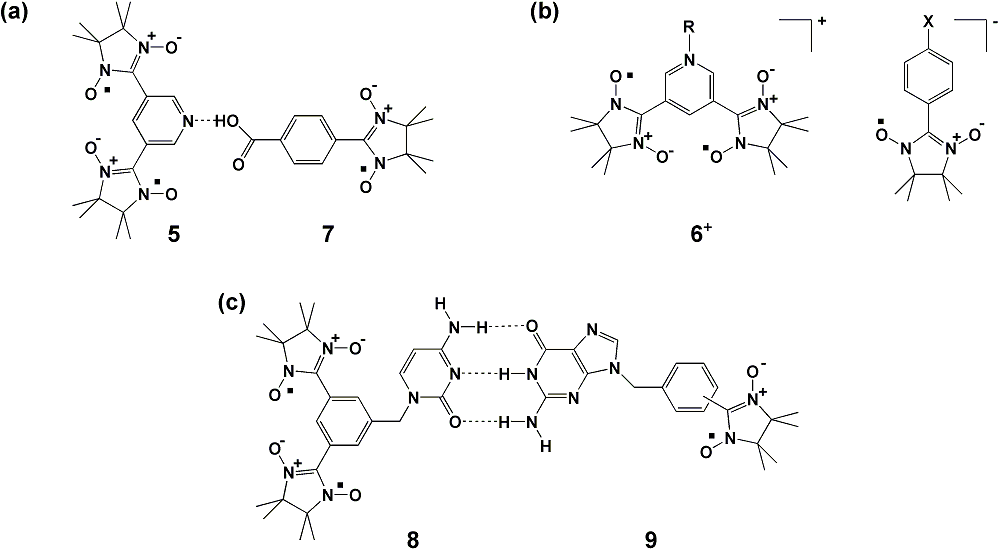
Scheme 2. Model
compounds for (a) hydrogen-bonded complex (X = phenolate, carboxylate),
(b) organic-salt ferrimagnets, and (c) bio-inspired ferrimagnets.
(d) Generalization of
Ferrimagnetism: Approaches towards Molecular Devices.
The deviation of spin value <Sb2>
for
organic
biradicals from that of an isolated molecule has been found for
ground-state
singlet (Sb = 0) biradicals as well. In
non-quantum terms the ground-state singlet biradicals would seemingly
have no contribution to the magnetization. An Sb = 0
is, however, not a good quantum number when the biradical is
interacting
with neighboring molecules. Theoretical investigation for molecular
assemblages of the ground-state singlet biradicals and monoradicals
indicate that the biradicals recover their contribution to the bulk
magnetization; <Sb 2> >
0, leading to a generalized ferrimagnetic high-spin state (Fig.4).
Subtle modification of the intermolecular interactions results in
possible switching between the ferrimagnetic high-spin and quantum
mechanically disordered low-spin states. This switching gives a new
category of magnetic materials, leading to spin-mediated molecular
devices. Model compounds for generalized ferrimagnets 11-13
have been
synthesized, which are based on single-component ferrimagnet approach,
and their magnetic properties are elucidated on the basis of the
crystal structures
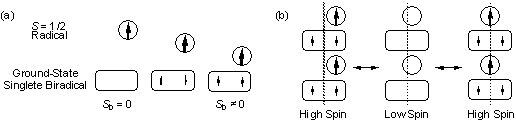
Fig.4. Schematic drawings of
the recovery of magnetic moment at the ground-state singlet biradical
(a) and the resulting switching of bulk magnetism (b).
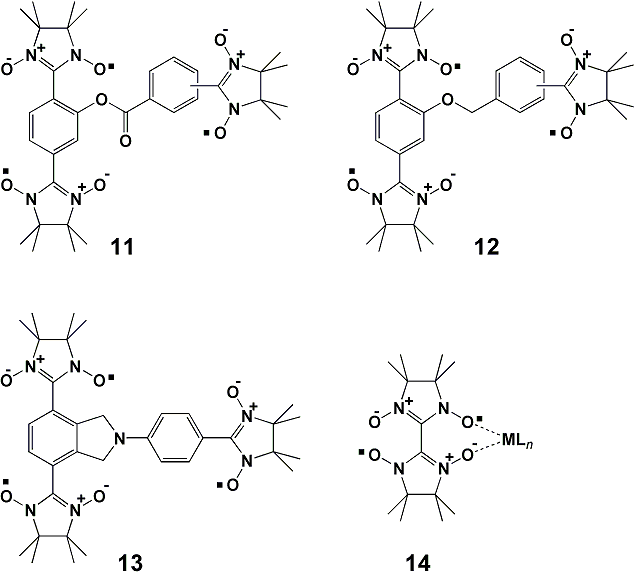
Scheme 3. Model compounds for generalized ferrimagnets.
2. Selected Publications
1. "Magnetic Phase Transition in a Heteromolecular Hydrogen-Bonded
Complex of Nitronylnitroxide Radicals", K. Hayakawa, D. Shiomi,
T. Ise, K. Sato, and T. Takui, J. Phys. Chem. B, 109,
9195-9197 (2005).
2. "Theoretical Study on Spin Alignments in Ferromagnetic Heterospin
Chains with Competing Exchange Interactions: A Generalized
Ferrimagnetic System Containing Ground-State Singlet Biradicals", K.
Maekawa, D. Shiomi, T. Ise, K. Sato, and T. Takui, J. Phys.
Chem. B, 109, 9299-9304 (2005).
3. "Magnetic Ordering in a Genuine Organic Crystal of Triangular
Antiferromagnetic Spin Units", K. Takeda, Y. Yoshida, Y. Inanaga, T.
Kawae, D. Shiomi, T. Ise, M. Kozaki, K. Okada, K. Sato, and T.
Takui, Phys. Rev. B, 72, 24435/1-6 (2005).
4. "Syntheses, Crystal Structures and Magnetic Properties of Nitronyl
Nitroxide Triradicals Composed of Ground-State Singlet Biradicals and
Monoradicals: Spin Clusters in the Crystal", T. Ise, D. Shiomi,
K. Sato, and T. Takui, Chem. Mater., 17, 4486-4492
(2005).
5. "Exchange Interaction in Covalently-Bonded Biradical-Monoradical
Composite Molecules", K. Maekawa, D. Shiomi, T. Ise, K. Sato,
and T. Takui, J. Phys. Chem. B, 109, 3303-3309 (2005).
6. "Cytosine-Substituted Nitronylnitroxide Radical: A Key Component for
Bio-Inspired Molecule-Based Magnetics", D. Shiomi, M. Nozaki,
T. Ise, K. Sato, and T. Takui, J. Phys. Chem. B, 108,
16606-16608 (2004).
7. "Stable Iminonitroxide Biradical in the Triplet Ground State", K.
Hayakawa, D. Shiomi, T. Ise, K. Sato, and T. Takui, Chem.
Lett., 33, 1494-1495 (2004).
8. "A Molecular Quantum Description of Spin Alignments in
Molecule-Based Ferrimagnets: Numerical Calculations of Thermodynamic
Properties", D. Shiomi, K. Sato and T. Takui, J. Phys.
Chem. A, 106, 2096-2103 (2002).
9. "Quantum Ferrimagnetism Based on Organic Biradicals with a Spin-0
Ground State: Numerical Calculations of Molecule-Based Ferrimagnetic
Spin Chains", D. Shiomi, K. Sato, and T. Takui, J. Phys.
Chem. B, 105, 2932-2938 (2001).
10. "Single-Component Molecule-Based Ferrimagnetics", D. Shiomi,
T. Kanaya, K. Sato, M. Mito, K. Takeda, and T. Takui, J. Am. Chem.
Soc., 123, 11823-11824 (2001).
|
